1 概述
汉德-许勒尔-克思斯琴病(Hand-Schüller-Christian disease)又称亚急性或慢性分化型组织细胞增多病,它比急性分化型组织细胞增多病(Letterer-Siwe disease)更易引起骨破坏,但比Letterer-Siwe病预后好。现常常将两种病变视为同一种疾病,因为它们具有共同的组织病理学特点,只不过发病年龄不同,临床表现有差异,有急性和亚急性之分,预后也不同。治疗可坚持口服泼尼松,对孤立的病灶、骨性病变和突眼可行小剂量的放射治疗,也可用长春新碱、环磷酰胺、甲氨蝶呤等药物进行化学治疗。
7 汉德-许勒尔-克思斯琴病的病因
汉德-许勒尔-克思斯琴病病因不明,可能与免疫功能紊乱有关。
9 汉德-许勒尔-克思斯琴病的临床表现
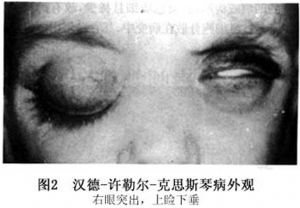
汉德-许勒尔-克思斯琴病多发性在3岁以上的儿童,成人少见发生。患者面部、眼睑、躯干、会阴和腋下皮肤发生溃疡或黄色瘤,口腔黏膜溃疡。肺门和肺间质因组织细胞和炎性细胞浸润而发生纤维化,可引起右心衰竭。垂体或下丘脑受累而发生尿崩症。颅骨、颅底骨、蝶鞍、上下颌骨、骨盆、股骨、肋骨和肱骨均可受累,特别是局限性、大小不等、边界不规则、边缘清楚,无硬化现象的缺损区,形似地图,故称地图样骨缺损(图1)。眼眶外壁和眶顶骨受破坏,眼眶软组织可能受累产生眼球突出,但眼球突出的真正原因不明(图2)。典型病例出现颅骨地图样缺损、突眼和尿崩症三联征,但非典型病例三大特征不会同时出现,或仅有其中一或二个症状,尿崩症是病变后期的并发症。

10 汉德-许勒尔-克思斯琴病的并发症
全身多处溃疡形成。
11 实验室检查
在非典型病变应进行实验室检查,可发现骨髓病性贫血,白细胞和血小板减少。骨髓内发现脂性巨细胞、淋巴细胞、嗜酸性细胞,必要时做活体组织检查以确定诊断。确定有无骨髓性贫血。
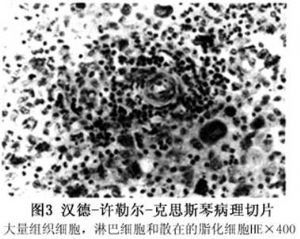
病理学检查:可见成熟的组织细胞核呈卵圆形,核膜轻度凹陷,染色质粗而较稠密,细胞质嗜酸性明显。在成熟组织细胞的背景中出现单核炎性细胞,有较多的嗜酸性细胞,散在淋巴细胞和浆细胞,也可见多核巨细胞。组织细胞和多核巨细胞吞噬脂质使其脂化(图3)。病变可因纤维结缔组织增生硬化而愈合。
15 汉德-许勒尔-克思斯琴病的治疗
可用泼尼松1mg/kg,每天口服,2个月后逐渐减量。对孤立的病灶、骨性病变和突眼可行小剂量的放射治疗,也可用长春新碱、环磷酰胺、甲氨蝶呤等药物进行化学治疗。最好由小儿肿瘤专家配合实施治疗。
16 预后
预后取决于患者年龄,临床表现和适当的治疗,病死率50%左右。