2 附录Ⅵ A 氮测定法(半微量法)
本法系依据含氮有机物经硫酸消化后,生成硫酸铵,硫酸铵被氢氧化钠分解释放出氨,后者借水蒸气被蒸馏入硼酸液中生成硼酸铵,最后用强酸滴定,依据强酸消耗量可计算出供试品的氮含量。
2.1 试剂
(1)消化剂 称取硫酸铜(CuSO4·5H2O10g、硫酸钾100g,置乳钵内,共同研细,混匀。
(2)混合指示液 0.2%溴甲酚绿乙醇溶液5份与0.1%甲基红乙醇溶液2份混合配成。
(3)2%硼酸吸收液 称取硼酸20g,加水溶解并稀释至1000ml,加混合指示液10ml,混匀。
(4)硫酸滴定液(0.05mol/L) 量取硫酸3ml,缓缓注入适量水中,冷却至室温,加水稀释至1000ml,摇匀。
取在270~300℃干燥至恒重的基准无水碳酸钠约0.15g,精密称定,加水50ml使溶解,加甲基红-溴甲酚绿混合指示剂10滴,用本液滴定至溶液由绿色转变为紫红色时,煮沸2分钟,冷却至室温,继续滴定至溶液由绿色变为暗紫色。每1ml硫酸滴定液(0.05mol/L)相当于5.30mg的无水碳酸钠。根据本液的消耗量与无水碳酸钠的取用量,算出本液的浓度,即得。
(5)硫酸滴定液(0.005mol/L) 精密量取硫酸滴定液(0.05mol/L) 100ml,置1000ml量瓶中,加水稀释至刻度,摇匀。
2.2 测定法
精密量取一定体积的供试品(约相当于含氮量1.0~2.0mg),置凯氏定氮瓶中,加消化剂约0.3g,硫酸1ml消化至澄明,呈蓝绿色,继续消化约60分钟。量取2%硼酸吸收液10ml置100ml锥形瓶内,将凯氏蒸馏器冷凝管末端浸入硼酸吸收液内,将消化好的供试品移入凯氏蒸馏器内,用水洗定氮瓶3~4次,将洗液移人蒸馏器内,再加入50%氢氧化钠溶液5ml,然后进行蒸馏,待接收液总体积约35~50ml,将冷凝管末端移出液面,使蒸汽继续冲洗约1分钟,用水淋洗尖端后停止蒸馏。接收液用硫酸滴定液(0.005mol/L)进行滴定,至溶液由蓝绿色变为灰紫色,并将滴定的结果用空白试验校正。
按下式计算:
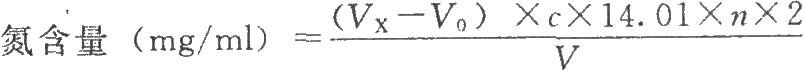
式中VX为供试品消耗酸滴定液的体积,ml;
n为供试品的稀释倍数;
V为供试品溶液的体积,ml;
14.01为氮的相对原子质量。
2.3 【附注】
(1)蒸馏前应蒸洗蒸馏器15分钟以上。
(3)也可用全自动凯氏定氮仪测定,按仪器使用说明书进行操作。
3 附录Ⅵ B 蛋白质测定法
3.1 第一法 凯氏定氮法
3.1.1 1.钨酸沉淀法
本法系通过测定供试品的总氮含量以及经钨酸沉淀去除蛋白质的供试品滤液中的非蛋白氮含量,计算出蛋白质的含量。
供试品溶液的制备
(1)总氮测定溶液的制备 精密量取供试品1ml,用生理氯化钠溶液准确稀释至每1ml含氮量约1mg。
(2)非蛋白氮测定溶液的制备 精密量取供试品2ml,加水14ml、10%钨酸钠溶液2ml、硫酸溶液(1.86→100) 2ml,摇匀,静置30分钟过滤,取滤液测定。
测定法 精密量取总氮测定溶液1ml,置凯氏定氮瓶中,照氮测定法(2010年版药典三部附录Ⅵ A)测定供试品中总氮含量,同时做空白对照。
精密量取非蛋白氮测定溶液5ml,置凯氏定氮瓶中,照氮测定法(2010年版药典三部附录Ⅵ A)测定供试品中非蛋白氮含量。按下式计算:

式中CTN为供试品溶液总氮含量,mg/ml;
CNPN为供试品溶液非蛋白氮含量,mg/ml;
V0为空白试验消耗酸漓定液的体积,ml;
CPN为供试品蛋白氮含量,mg/ml;
n为供试品的稀释倍数;
V1为供试品总氮测定溶液的体积,ml;
V2为供试品非氮测定溶液的体积,ml;
14.01为氮的相对原予质量;
【附注】
(1)供试品蛋白质含量如超过10%(g/ml),除蛋白质时应适当加大供试品稀释倍数,10%钨酸钠溶液及硫酸溶液用量相应地按比例增加,使溶液中的钨酸浓度仍保持1%。
(2)总氮测定和非蛋白氮测定可用同一空白对照。
3.1.2 2.三氯乙酸沉淀法
本法系将供试品经三氯乙酸沉淀,通过测定该沉淀中的蛋白氮含量,计算出蛋白质的含量。
测定法 精确量取适宜体积的供试品(每1ml含蛋白质4~10mg)于适宜的尖底离心管中,加等体积的12%三氯乙酸(12→100)混匀,静置30分钟后,以每分钟4000转离心,弃上清液,用约3ml水分数次将沉淀洗入凯氏定氮瓶中,照氮测定法(2010年版药典三部附录Ⅵ A)测定供试品中总氮含量,同时做空白对照。
按下式计算:
供试品蛋白质含量(mg/ml) 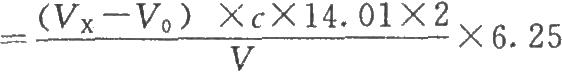
式中VX为供试品消耗酸滴定液的体积,ml;
V为供试品的体积,ml。
14.01为氮的相对原子质量;
3.2 第二法 Lowry法
本法用于微量蛋白质的含量测定。蛋白质在碱性溶液中可形成铜-蛋白质复合物,此复合物加入酚试剂后,产生蓝色化合物,该蓝色化合物在波长650nm处的吸光度与蛋白质含量成正比,根据供试品的吸光度,计算供试品的蛋白质含量。
3.2.1 试剂
(1)酚试剂 称取钨酸钠(Na2WO4·2H2O) 100g、钼酸钠(Na2MnO4·2H2O) 25g,置1500ml蒸馏瓶中,加入700ml水、85%磷酸50ml、盐酸100ml,上连回流管(使用木塞或锡纸包裹的橡皮塞)微沸回流10小时。取下回流管,加入硫酸锂150g、水50ml、溴液几滴,煮沸约15分钟,驱除过量的溴。冷却,加水至1000ml,过滤,为酚试剂贮备液。
酚试剂贮备液用氢氧化钠滴定液(0.5mol/L)测定酸浓度,然后加水稀释至相当于1mol/L盐酸浓度。
(2)碱性铜溶液 量取0.1mol/L酒石酸钾溶液与0.04mol/L硫酸铜溶液各0.5ml,4%碳酸钠溶液与0.8%氢氧化钠溶液各25ml,摇匀,即得。本液应临用配制。
(3)氢氧化钠滴定液(0.5mol/L)的配制及标定取氢氧化钠适量,加水搅拌使溶解成饱和溶液,冷却后,置聚乙烯塑料瓶中,静置数日,澄清后,量取澄清的氢氧化钠饱和溶液28ml,加新煮沸后冷却的水,使成1000ml,摇匀。
取在105℃干燥至恒重的基准邻苯二甲酸氢钾约3g,精密称定,加新沸过的冷水50ml,振摇,使其尽量溶解;加酚酞指示剂2滴,用本液滴定;在接近终点时,应使邻苯二甲酸氢钾完全溶解,滴定至溶液显粉红色。每1ml氢氧化钠滴定液相当于102.1mg的邻苯二甲酸氢钾。根据本液的消耗量与邻苯二甲酸氢钾的取用量,算出本液的浓度。
3.2.2 标准蛋白质溶液的制备
用水将蛋白质标准品定量稀释至每1ml含1mg,作为贮备液。精密量取贮备液2.5ml置25ml量瓶中,用水稀释至刻度,摇匀,即为每1ml含100μg的标准蛋白质溶液。
3.2.3 测定法
精密量取一定体积供试品(含蛋白质50μg左右)置试管内,加水至1ml,加碱性铜溶液5ml,摇匀,室温放置10分钟,快速加入酚试剂0.5ml,摇匀,室温放置30分钟,显色后,照紫外一可见分光光度法(2010年版药典三部附录Ⅱ A),在波长650nm处测定吸光度(呈色后,如发现浑浊,经每分钟3000转离心15分钟后,取上清液测定)。精密量取标准蛋白质溶液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml分别置于试管中,自“加水至1ml”起,同法操作。准确量取水1ml,自“加碱性铜溶液5ml”起,同法操作,作空白对照。以标准品的蛋白质浓度对其相应吸光度作直线回归,求得直线回归方程;将测得供试品的吸光度代入直线回归方程,即得供试品的蛋白质含量。
3.3 第三法 双缩脲法
本法系依据蛋白质肽键在碱性溶液中与Cu2+形成紫红色络合物,其颜色深浅与蛋白质含量成正比,利用标准蛋白质溶液作对照,采用紫外一可见分光光度法测定供试品蛋白质含量。
3.3.1 试剂
双缩脲试剂 取硫酸铜(CuSO4·5H2O)3.0g、酒石酸钾钠(KNaC4H4O6·4H2O)9.0g、碘化钾5.0g、氢氧化钠24g,加水溶解并稀释至1000ml,摇匀,即得。
3.3.2 标准蛋白质溶液的制备
3.3.3 供试品溶液的制备
精密量取适量的供试品,加水制成每1ml约含50mg的溶液。
3.3.4 测定法
分别精密量取供试品溶液与标准蛋白质溶液各0.05ml置玻璃试管中,分别加双缩脲试剂4.0ml,混匀,置37℃水浴中30分钟,照紫外-可见分光光度法(2010年版药典三部附录Ⅱ A),在波长540nm处测定吸光度。另精密量取水0.05ml,自“加双缩脲试剂4.0ml”起,同法操作,作为空白对照。
按下式计算:
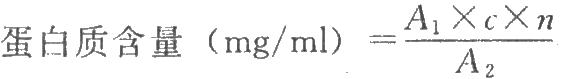
式中A1为供试品溶液的吸光度;
n为供试品稀释倍数。
【附注】 本法的测定范围为1~10mg。
4 附录Ⅵ C 唾液酸测定法(间苯二酚显色法)
本法系用酸水解方法将结合状态的唾液酸变成游离状态,游离状态的唾液酸与间苯二酚反应生成有色化合物,再用有机酸萃取后,测定唾液酸含量。
4.1 唾液酸对照品溶液(200Vg/ml)的制备
精密称取唾液酸对照品10.52mg(1μg唾液酸相当于3.24nmol),置10ml量瓶中,加水溶解并稀释至刻度,混匀,即为唾液酸贮备液(1mg/ml),按一次使用量分装,-70℃贮存,有效期1年。仅可冻融1次。4℃保存使用期为2周。精密量取唾液酸贮备液1ml,置5ml量瓶中,加水至刻度,即为每1ml含200μg的唾液酸对照品溶液,用前配制。
4.2 测定法
取供试品适量,加水稀释至蛋白质浓度约为每1ml含0.2~0.4mg,作为供试品溶液。按下表取唾液酸对照品溶液、水及供试品溶液于10ml玻璃试管中,混匀,每管再加入间苯二酚-盐酸溶液(分别量取2%间苯二酚溶液2.5ml、0.1mol/L硫酸铜溶液62.5μl、25%盐酸溶液20ml,加水稀释至25ml,混匀。试验前4小时内配制)1ml,,加盖,沸水煮沸30分钟(水浴面高于液面约2cm),取出置冰浴中3分钟(同时振摇)后,每管加乙酸丁酯-丁醇液(取4体积乙酸丁酯与1体积丁醇混匀,室温下保存,12小时内使用)2ml,充分混匀,室温放置10分钟,照紫外-可见分光光度法(2010年版药典三部附录Ⅱ A),在波长580nm处测定吸光度。
以唾液酸对照品溶液的浓度对其相应的吸光度作直线回归(相关系数应不低于0.99),由直线回归方程求出5μg唾液酸的吸光度,再按下式计算:
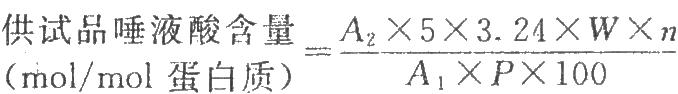
式中A1为5μg唾液酸的吸光度;
A2为供试品的吸光度;
n为供试品稀释倍数;
P为供试品蛋白质含量,μg/μl;
W为1nmol促红素的量[1],相当于30.6μg。
5 附录Ⅵ D 乙醇残留量测定法(康卫皿扩散法)
本法系依据乙醇在饱和碳酸钠溶液中加热逸出,被重铬酸钾-硫酸溶液吸收后呈黄绿色至绿色,用比色法测定血液制品中乙醇残留量。
5.1 测定法
在康卫皿外圈的凸出部位均匀涂抹凡士林,准确量取重铬酸钾-硫酸溶液(称取重铬酸钾3.7g,加水150ml,充分溶解后缓慢加入硫酸280ml,放冷,加水至500ml,摇匀)2.0ml加入内圈中,量取饱和碳酸钠溶液[称取碳酸钠(Na2CO3·10H2O)适量,加等重量的水,充分摇匀,取上清液] 1.5ml和精密量取的供试品溶液1.5ml,加入外圈中,立即加盖玻璃板(粗糙面向下)密封扩散皿,摇匀,80℃反应30分钟后,取内圈溶液,照紫外-可见分光光度法(2010年版药典三部附录Ⅱ A),在波长650nm处测定吸光度(A1)。精密量取无水乙醇适量,加水制成每1ml中含乙醇0.25mg的溶液,即为对照品溶液。精密量取对照品溶液1.5ml替代供试品,同法操作,测定吸光度(A2)。A1不得大于A2。
6 附录Ⅵ E 组胺人免疫球蛋白中游离磷酸组胺测定法
本法系依据磷酸组胺与邻苯二甲醛在碱性条件下生成荧光衍生物,以此测定组胺人免疫球蛋白中游离磷酸组胺含量。
6.1 磷酸组胺对照品溶液的制备
取磷酸组胺对照品7mg,精密称定,置25ml量瓶中,用0.1mol/L盐酸溶液溶解并稀释至刻度,摇匀,作为磷酸组胺贮备液,-20℃贮存备用。试验当天准确量取磷酸组胺贮备液0.1ml,置100ml量瓶中,用0.1mol/L盐酸溶液稀释至刻度,即为磷酸组胺对照品溶液。
6.2 供试品溶液的制备
量取供试品0.5ml,加水1.2ml,混匀,加25%三氯乙酸溶液0.3ml,混匀,以每分钟4000转离心10分钟,取上清液,即为供试品溶液。
6.3 测定法
量取供试品溶液1.6ml置试管中,加氯化钠1.5g,再加正丁醇4.0ml、2.5mol/L氢氧化钠溶液0.2ml,立即混匀5分钟,静置后,取出正丁醇相3.6ml加到已装有0.1mol/L盐酸溶液1.2ml和正庚烷2.0ml的试管内,振荡5分钟,弃有机相,量取盐酸相1.0ml,加入等体积水,再加0.4mol/L氢氧化钠溶液0.5ml,混匀并迅速加入0.1%邻苯二甲醛-甲醇溶液0.1ml,立即混匀,置21~22℃10分钟,加0.5mol/L盐酸溶液0.5ml终止反应,取终止反应后的溶液200μl,加入酶标板孔中,用荧光酶标仪,在激发波长350nm和发射波长450nm处测荧光强度。
准确量取磷酸组胺对照品溶液1.0ml、0.8ml、0.6ml、0.4ml、0.2ml、0.1ml、0.05ml、0.025ml,分别置试管中,各以0.1mol/L盐酸溶液补足至1.0ml;向各管中加水0.5ml、25%三氯乙酸溶液0.1ml,混匀,加氯化钠1.5g,自“加正丁醇4.0ml”起,同法操作。
以磷酸组胺对照品溶液的浓度对其相应的荧光强度作直线回归,将供试品溶液的荧光强度代入直线回归方程,求出供试晶溶液碱基含量(G),按下式计算:
供试品游离磷酸组胺含量(ng/ml)=G×2.76×2.5
【附注】 磷酸组胺分子量为307.148,对照品溶液浓度按碱基计,碱基与磷酸组胺分子量比为1:2.76。式中2.5为供试品稀释倍数。
7 附录Ⅵ F O-乙酰基测定法
7.1 试剂
(1) 2mol/L盐酸羟胺溶液 称取盐酸羟胺13.9g,加水溶解并稀释至100ml,冷处保存。
(2)3.5mol/L氢氧化钠溶液 称取氢氧化钠14.0g,加水使溶解并稀释至100ml。
(3) 4mol/L盐酸溶液 量取盐酸33.3ml,加水稀释至100ml的溶液。
(4)0.37mol/L三氯化铁一盐酸溶液 称取三氯化铁(FeCl3·6H20) 10.Og,加0.1mol/L盐酸溶液溶解并稀释至100ml。
(5)碱性羟胺溶液 量取等体积的盐酸羟胺溶液(2mol/L)与氢氧化钠溶液(3.5mol/L)混合。3小时内使用。
7.2 对照品溶液的制备
精密称取已干燥至恒重的氯化乙酰胆碱22.7mg(或溴化乙酰胆碱28.3mg),置50ml量瓶中,加0.001mol/L醋酸钠溶液(pH4.5)溶解并稀释至刻度,摇匀。
7.3 供试品溶液的制备
取供试品,用水稀释成O-乙酰基浓度为0.5~2.5mmol/L的溶液。
7.4 测定法
精密量取氯化乙酰胆碱(或溴化乙酰胆碱)对照品溶液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml,分别置试管中,补加水至1ml,加新鲜配制的碱性羟胺溶液2ml,摇匀,于室温放置4分钟,加4mol/L盐酸1ml,调pH值至1.2±0.2,摇匀,加0.37moI/L三氯化铁-盐酸溶液1ml,摇匀,照紫外-可见分光光度法(2010年版药典三部附录Ⅱ A),在波长540nm处测定吸光度。另精密量取上述相应的系列对照品溶液,自“补加水至1ml”起,除加酸与加碱性羟胺的次序颠倒外,同法操作,用作对应的空白对照。
精密量取供试品溶液1ml置试管中,自“加新鲜配制的碱性羟胺溶液2ml”起,同法操作;另取供试品溶液1ml,与对照品溶液的空白对照同法操作,用作供试品的空白对照。
将标准管各吸光度分别减去相应的空白对照管的吸光度,以标准管中所含的对照品溶液的体积对其相应的吸光度作直线回归,将供试品的吸光度减去相应的空白对照管的吸光度后代入直线回归方程,计算出每1ml供试品相当于对照品溶液的体积(V,ml)。
供试品中O-乙酰基含量(mmol/L)=V×2.5
8 附录Ⅵ G 聚乙二醇残留量测定法
本法系依据聚乙二醇与钡离子和碘离子形成复合物(1:1).用比色法测定聚乙二醇含量。
8.1 测定法
取供试品适量,用水稀释,使蛋白质浓度不高于1%,即为供试品溶液。精密量取供试品溶液1.0ml,加入0.5mol/L高氯酸溶液5.0ml,混匀,室温放置15分钟,以每分钟4000转离心10分钟。取上清液4ml,加入氯化钡溶液(称取氯化钡5g,加水溶解至100ml)1.0ml和0.1mol/L碘溶液(称取碘化钾2.0g,加少量水溶解,然后加碘1.3g,再加水至50ml,摇匀)0.5ml,混匀,室温反应15分钟,照紫外-可见分光光度法(2010年版药典三部附录Ⅱ A),在波长535nm处测定吸光度;同时以1ml水代替供试品溶液,同法操作,即为空白对照。
另精密称取聚乙二醇(分子量4000或6000)适量,加水溶解,并制成每1ml含聚乙二醇100μg的溶液,即为聚乙二醇对照品贮备液。
取按下表制备的每1ml含10~50μg的聚乙二醇对照品溶液1.0ml,加入0.5mol/L高氯酸溶液5.0ml,混匀,自“室温放置15分钟”起,同法操作。
以聚乙二醇对照品溶液的浓度(μg/ml)对其相应的吸光度作直线回归,将供试品溶液吸光度代入直线回归方程,计算出供试品溶液聚乙二醇含量F(μg/ml)。
按下式计算:
供试品聚乙二醇含量(g/L)=F×n×10-3
n为供试品稀释倍数。
8.2 【附注】
(1)整个比色过程应在试剂加入后的15~45分钟内完成,否则将要影响结果。
9 附录Ⅵ H 聚山梨酯80残留量测定法
本法系依据聚山梨酯80中的聚乙氧基(Polyethoxylated)和铵钴硫氰酸盐反应形成蓝色复合物,可溶于二氯甲烷,用比色法测定聚山梨酯80含量。
9.1 测定法
量取供试品1.0ml于离心管中,加乙醇-氯化钠饱和溶液5ml,摇匀,以每分钟3000转离心10分钟,取上清液,再用乙醇-氯化钠饱和溶液1.0ml小心冲洗管壁,洗液与上清液合并,以每分钟3000转离心10分钟,上清液置55℃水浴中,用空气吹扫法将其浓缩至0.1~0.5ml,加1ml水溶解。准确加入二氯甲烷2.0ml、硫氰钴铵溶液(称取硝酸钴6.0g、硫氰酸铵40.0g,加水溶解并稀释至200ml)3.0ml,加塞,混匀,室温放置1.5小时,每15分钟振荡1次,测定前静置半小时,弃上层液,照紫外-可见分光光度法(2010年版药典三部附录Ⅱ A),在波长620nm处测定下层二氯甲烷液的吸光度。用二氯甲烷作空白对照。
精密量取聚山梨酯80对照品溶液(取聚山梨酯80约100mg,精密称定,加水溶解后置100ml量瓶中,加水稀释至刻度)0μl、10μl、25μl、50μl、75μl、100μl,加入预先加入1ml水的离心管中混匀,准确加入二氯甲烷2.0ml、硫氰钴铵溶液3.0ml,加塞,混匀,自“室温放置1.5小时”起,同法操作。
以上述聚山梨酯80系列浓度(μg/ml)对其相应的吸光度作直线回归,相关系数应不低于0.98,将供试品吸光度代入直线回归方程,求得供试品聚山梨酯80含量(μg/ml)
10 附录Ⅵ I 戊二醛残留量测定法
本法系依据戊二醛与2,4-二硝基苯肼反应生成正戊醛二硝基苯肼,用高效液相色谱法,测定供试品中戊二醛含量。
照高效液相色谱法(2010年版药典三部附录Ⅲ B)测定。
10.1 色谱条件
用十八烷基硅烷键合硅胶填充剂(SG120,S-5μm,直径4.6mm,长250mm);以70%乙腈溶液为流动相;流速为每分钟1.2ml;检测波长为360nm;记录时间为30分钟。
10.2 测定法
取戊二醛对照品适量,精密称定.加水溶解并定量稀释成每1ml中约含10μg的溶液;精密量取该溶液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml.分别置试管中,各加水至1.0ml,精密加流动相1ml与2,4-二硝基苯肼溶液(称取2,4-二硝基苯肼2.4g,加30%高氯酸溶液,溶解成100ml)0.1ml,立即于混合器上混匀,用0.45μm膜滤过。另取供试品适量,以每分钟3000转离心10分钟,精密量取上清液1ml,自“精密加流动相1ml”起,同法操作。分别精密量取对照品溶液与供试品溶液各10μl,注入液相色谱仪,记录色谱图。
以戊二醛对照品溶液的浓度对其相应的峰面积作直线回归,求得直线回归方程,计算出供试品溶液中戊二醛含量。
10.3 【附注】
(1)配制戊二醛对照品溶液用的戊二醛用量0.1g(系经色谱纯度测定后折算其含量为100%)。
(2)直线回归相关系数应不低于0.99。
11 附录Ⅵ J 磷酸三丁酯残留量测定法
照气相色谱法(2010年版药典三部附录Ⅲ C)测定。
11.1 色谱条件与系统适用性试验
用酸改性聚乙二醇(20M)毛细管柱,1柱温140℃,气化室温度190℃,火焰离子化检测器或氮磷检测器,检测器温度210℃,载气(氮气)流速为每分钟60ml,或根据仪器选择检测条件。理论板数按磷酸三丁酯峰计算应不低于5000,磷酸三丁酯蜂与磷酸三丙酯峰的分离瘦应不小于1.5,磷酸三丁酯对照品溶液连续进样5次,所得磷酸三丁酯峰与磷酸三丙酯峰面积之比的相对标准偏差(RSD)应不大于5%。内标溶液的制备 取磷酸三丙酯适量,用正已烷溶解并定量稀释制成每1ml中约含400μg的溶液。
11.2 测定法
精密量取供试品3ml,置具塞玻璃离心管中,精密加内标溶液50μl与1.5nmol/L高氯酸溶液0.75ml,振荡1分钟;置37℃水浴保温10分钟后,再加正己烷4ml,振荡2分钟;以每分钟2000转离心20分钟,小心吸取上层正己烷,用空气流将其浓缩至约0.2ml(不能加热),取0.1μl注入气相色谱仪。另取磷酸三丁酯对照品适量,精密称定,加正已烷溶解并定量稀释制成每1ml中约含600μg的溶液;精密量取该溶液10μl、20μl、40μl、60μl、80μl,分别置已精密加水3ml的具塞玻璃离心管中,再向各对照品管精密加内标溶液50μl,自“振荡1分钟”起,同法操作。以各磷酸三丁酯对照品峰面积与内标峰面积比,对磷酸三丁酯对照品溶液浓度作直线回归,求得直线回归方程,计算出供试品中磷酸三丁酯含量(μg/ml)。
11.3 【附注】
(1)对照品溶液与供试品溶液的溶剂挥发的速度应尽量保持一致;若离心后,乳化仍未完全破除,可在振荡器上稍微振摇一下,再离心1次。
(2)直线回归相关系数应不低于0.99。
12 附录Ⅵ K 辛酸钠测定法
本法系用气相色谱法测定供试品中辛酸钠含量。照气相色谱法(2010年版药典三部附录Ⅲ C)测定。
12.1 色谱条件与系统适用性试验
用酸改性聚乙二醇(20M)毛细管柱,柱温160℃,火焰离子化检测器,检测器温度230℃,气化室温度230℃,载气(氮气)流速为每分钟35ml。辛酸峰与庚酸峰的分离度应大于1.5,辛酸峰的拖尾因子应为0.95~1.20,辛酸对照品溶液连续进样5次,所得辛酸峰与庚酸峰面积之比的相对标准偏差(RSD)应不大于5%。
12.2 内标溶液的制备
12.3 测定法
取供试品,用水准确稀释成每升含蛋白质40~50g的溶液,即为供试品溶液。精密量取供试品溶液0.5ml,加内标溶液30μl与1.5mol/L高氯酸溶液0.2ml,于振荡器上混合1分钟,加三氯甲烷4ml,加盖,于振荡器上剧烈混合2分钟,以每分钟3000转离心20分钟,除去上层水相,小心将三氯甲烷层倾入10ml试管中,将三氯甲烷挥发至干,加三氯甲烷100μl溶解残渣,取0.1μl注入气相色谱仪。另取辛酸对照品约0.15g,精密称定,置10ml量瓶中,用三氯甲烷溶解并稀释至刻度,即为辛酸对照品溶液。精密量取辛酸对照品溶液10μl、20μl、30μl、40μl、50μl,各精密加内标溶液30μl,于振荡器上混合1分钟,加三氯甲烷4ml,将三氯甲烷挥发至干,各加三氯甲烷100μl溶解残渣,同法操作。
以各辛酸对照品溶液峰面积与内标峰面积比对各辛酸对照品溶液辛酸量(μg)作直线回归,求得直线回归方程,计算出供试品溶液辛酸绝对量(A),再按下式计算供试品辛酸钠含量:

B为取样量,即为0.5ml;
n为供试品稀释倍数;
c为供试品蛋白质含量,g/ml;
12.4 【附注】
(1)1mmol辛酸相当于1mmol辛酸钠。
(2)对照品溶液与供试品溶液的溶剂挥发的速度应尽量保持一致。
(3)直线回归相关系数应不低于0.99。
13 附录Ⅵ L 游离甲醛测定法
本法系依据品红亚硫酸在酸性溶液中能与甲醛生成紫色复合物,用比色法测定供试品中游离甲醛含量。
13.1 对照品溶液的制备
精密量取已标定的甲醛溶液适量,置500ml量瓶中,用水稀释至刻度,摇匀,制成0.05%甲醛对照品贮备液。
临用前,精密量取甲醛对照品贮备液10ml,置100ml量瓶中,加水稀释至刻度,摇匀,作为甲醛对照品溶液。测定法 精密量取供试品1ml,用水稀释至甲醛含量约为0.005%,即为供试品溶液。精密量取供试品溶液1ml,置50ml具塞试管中,加水4ml,加品红亚硫酸溶液10ml,混合酸溶液(量取水783ml,置烧杯内,缓缓注入盐酸42ml、硫酸175ml,混匀)10ml,摇匀,于25℃放置3小时,照紫外-可见分光光度法(2010年版药典三部附录Ⅱ A),在波长590nm处测定吸光度。
精密量取0.005%甲醛对照品溶液0.5ml、1.0ml、1.5ml、2.0ml,分别置。50ml具塞试管中,加水至5ml,自“加品红亚硫酸溶液10ml”起,同法操作。
以甲醛对照品溶液的浓度对相应的吸光度作直线回归,将供试品溶液的吸光度代入直线回归方程,计算供试品中的游离甲醛含量。
【附注】
(1)品红亚硫酸溶液的制备及二氧化硫含量的标定 称取碱性品红4.5g,于3000ml锥形瓶中,加水1500ml,振摇或加温使品红全部溶解,待冷后,加亚硫酸钠10g,摇匀,静置5~10分钟,再加入3nmol/L硫酸溶液40ml,摇匀,以橡皮塞塞紧瓶口,放置过夜,如有颜色,加骨炭5~10g迅速摇匀,以布氏漏斗快速抽滤,即得品红亚硫酸溶液。品红亚硫酸溶液中的SO2含量可控制在28~48mmol/L(SO2含量过多可通空气驱除,过少可通人SO2)。
二氧化硫(SO2)含量测定 量取品红亚硫酸溶液10ml于锥形瓶内,加水20ml,淀粉指示液5ml,用碘滴定液(0.05mol/L)滴定至呈浅蓝色,按下式计算SO2的含量;
SO2的含量(mmol/L)=50×V×c
式中 V为消耗碘滴定液(0.05mol/L)的体积,ml;
c为碘滴定液的浓度,mol/L。
(2)甲醛溶液的标定 取甲醛溶液约1.5ml,精密称定,置锥形瓶中,加水10ml、过氧化氢溶液25ml与溴麝香草酚蓝指示液2滴,滴加氢氧化钠滴定液(,1mol/L)至溶液显蓝色;再精密加入氢氧化钠滴定液(1mol/L)25ml,瓶口置一玻璃小漏斗,置水浴中加热15分钟,不时振摇,冷却,用水洗涤漏斗,加溴麝香草酚蓝指示液2滴,用盐酸滴定液(1mol/L)滴定至溶液显黄色,并将滴定的结果用空白试验校正。每1ml氢氧化钠滴定液(1mol/L)相当于30.03mg的甲醛。
(3)对照品溶液和供试品溶液与品红亚硫酸溶液的显色时间有时不一致,测定时,显色慢者应酌情早加品红亚硫酸溶液。
(4)供试品中如含有酚红,标准管中应予以校正。附录Ⅵ M 苯酚测定法本法系依据溴酸盐溶液与盐酸反应产生溴,遇苯酚生成三溴苯酚,过量的溴与碘化钾反应释出碘,析出的碘用硫代硫酸钠滴定液滴定,根据硫代硫酸钠滴定液的消耗量,可计算出供试品中苯酚的含量。
13.2 测定法
精密量取供试品1ml,置具塞锥形瓶中,加水50ml,精密加入0.02mol/L溴溶液(称取溴酸钾0.56g,加溴化钾3g,加水溶解并稀释至1000ml)15~25ml(供试品含苯酚量0.3%~0.5%时加25ml,小于0.3%则加15ml),沿瓶壁加入6mol/L盐酸溶液10ml,摇匀,密塞,在暗处放置30分钟后,加25%碘化钾溶液2ml于具塞锥形瓶颈口,稍启瓶塞,使流下,密塞,摇匀。以少量水洗瓶颈,用硫代硫酸钠滴定液(0.02mol/L)滴定至近终点时,加淀粉指示液约0.5ml,滴定至蓝色消失,并将滴定的结果用空白试验校正。
按下式计算:

【附注】
(1)硫代硫酸钠滴定液(0.1mol/L)的制备及标定 称取硫代硫酸钠26g与无水碳酸钠0.20g,加新沸过的冷水适量溶解并稀释至1000ml,摇匀,放置1个月后滤过。取在120℃干燥至恒重的基准重铬酸钾0.15g,精密称定,置碘瓶中,加水50ml溶解,加碘化钾2.0g,轻轻振摇使溶解,加稀硫酸(5.7→100) 40ml,摇匀,密塞;在暗处放置10分钟后,加水250ml稀释,用本液滴定至近终点时,加淀粉指示液(称取可溶性淀粉0.5g,加水5ml混悬后缓缓倾入100ml沸水中,随加随搅拌,继续煮沸2分钟,冷却,倾取上层清液。本液应临用配制)3ml,继续滴定至蓝色消失而显亮绿色,并将滴定的结果用空白试验校正。每1ml硫代硫酸钠滴定液(0.1mol/L)相当于4.903mg重铬酸钾。根据本液的消耗量与重铬酸钾的取用量,算出本液的浓度,即得。
(2)硫代硫酸钠滴定液(0.02mol/L)的制备 精密量取硫代硫酸钠滴定液(0.1mol/L)100ml,加水准确稀释至500ml,摇匀。
(3)可做限度测定。
14 附录Ⅵ N 间甲酚测定法
本法系依据4-氨基安替比林、铁氰化钾在碱性条件下与间甲酚反应生成一种红色物质,用比色法测定供试品中间甲酚含量。
14.1 测定法
精密量取一定体积的供试品,置试管中,定量稀释50倍,即为供试品溶液。量取供试品溶液1.0ml,加水5.0ml,混匀,依次加pH9.8缓冲液(称取无水碳酸钠6.36g、碳酸氢钠3.36g,加水溶解并稀释至800ml,用1mol/L盐酸调pH值至9.8后,再加水至1000ml)、0.3% 4-氨基安替比林溶液、1.2%铁氰化钾溶液及1mol/L磷酸二氢钾溶液各1.0ml,混匀,于室温避光放置10分钟,照紫外-可见分光光度法(2010年版药典三部附录Ⅱ A),在波长510nm处测定吸光度。
精密量取间甲酚对照品溶液(取间甲酚适量,精密称定,置量瓶中,加水溶解并稀释至每1ml含10μg)1.0ml、2.0ml、3.0ml、4.0ml、5.0ml、6.0ml,分别置试管中,加水补至6.0ml,自“依次加pH9.8缓冲液”起,同法操作,测定各管的吸光度。
以间甲酚对照品溶液的系列浓度对其相应的吸光度作直线回归,将供试品溶液的吸光度代入直线回归方程,得供试品的间甲酚含量(mg/ml)。
15 附录Ⅵ P 人血液制品中糖及糖醇测定法
照离子色谱法(2010年版药典三部附录Ⅲ E)测定。
15.1 色谱条件与系统适用性试验
用苯乙烯-二乙烯基苯共聚物为基质的阳离子交换色谱柱(H+),粒度9μm或8μm,内径7.8mm,柱长300mm;柱温50℃(测定蔗糖含量时,柱温为20~30℃);流动相为0.004mol/L硫酸溶液,流速为每分钟0.8ml;示差折光检测器。取2%麦芽糖1ml和1.5%磺基水杨酸1ml的混合物20μl,注入色谱柱,记录色谱图,麦芽糖与磺基水杨酸两峰间的分离度应大于1.5,拖尾因子按麦芽糖峰计算应为0.95~1.50。
15.2 对照品溶液的制备
(1)麦芽糖对照品溶液 分别取经减压干燥至恒重的麦芽糖对照品1.0g、2.0g、3.0g,精密称定,各置100ml量瓶中,分别加水溶解并稀释至刻度,摇匀,即得。
(2)葡萄糖对照品溶液 分别取经减压干燥至恒重的葡萄糖对照品0.5g、1.0g、1.5g,精密称定,各置100ml量瓶中,分别加水溶解并稀释至刻度,摇匀,即得。
(3)山梨醇对照品溶液 分别取经减压干燥至恒重的山梨醇对照品0.5g、1.0g、1.5g,精密称定,各置100ml量瓶中,分别加水溶解并稀释至刻度,摇匀,即得。
(4)蔗糖对照品溶液 分别取经减压干燥至恒重的蔗糖对照品1.0g、2.0g、3.0g,精密称定,各置100ml量瓶中,分别加水溶解并稀释至刻度,摇匀,即得。
15.3 供试品溶液的制备
精密量取供试品1ml,加1.5%磺基水杨酸4.0ml,混匀,室温放置至少2小时,以每分钟3000转离心10分钟,取上清液,即得。
15.4 测定法
精密量取对照品溶液与供试品溶液,分别注入液相色谱仪,记录色谱图;进样量为20μl。
以各对照品溶液浓度(g/L)对其相应的的峰面积作直线回归,求得直线回归方程,计算出供试品溶液中糖或糖醇含量(A),再按下列公式计算:
供试品糖或糖醇含量(g/L)=A×n
n为供试品稀释倍数。
【附注】
(1)根据供试品的糖含量,对照品和供试品的取量可做适当调整。
(2)直线回归相关系数应不低于0.999。
(3)不同厂家的阳离子交换色谱柱(H+)的流速、流动相、柱温等会有所不同,可根据色谱柱介绍对色谱条件进行适当调整。
16 附录Ⅵ Q 人血白蛋白多聚体测定法
照分子排阻色谱法(2010年版药典三部附录Ⅲ D)测定。
16.1 色谱条件与系统适用性试验
用亲水硅胶高效体积排阻色谱柱(SEC,排阻极限300kD,粒度10μm),柱直径7.5mm,长60cm;以含1%异丙醇的pH7.0、0.2mol/L磷酸盐缓冲液(量取0.5mol/L磷酸二氢钠200ml、0.5mol/L磷酸氢二钠420ml、异丙醇15.5ml及水914.5ml,混匀]为流动相;检测波长为280nm;流速为每分钟0.6ml。取每1ml含蛋白质12mg的人血白蛋白溶液20μl.注入色谱柱,记录色谱图,人血白蛋白单体峰与二聚体峰间的分离度应大于1.5,拖尾因子按人血白蛋白单体峰计算应为0.95~1.40。
16.2 测定法
17 附录Ⅵ R 人免疫球蛋白类制品IgG单体加二聚体测定法
本法系用分子排阻色谱法测定人免疫球蛋白类制品IgG单体加二聚体含量。
照分子排阻色谱法(2010年版药典三部附录Ⅲ D)测定。
17.1 色谱条件与系统适用性试验
用亲水硅胶高效体积排阻色谱柱(SEC,排阻极限300kD,粒度10μm),柱直径7.5mm,长60cm。以含1%异丙醇的pH7.0、0.2mol/L磷酸盐缓冲液(量取0.5mol/L磷酸二氢钠200ml、0.5mol/L磷酸氢二钠420ml、异丙醇15.5ml及水914.5ml,混匀]为流动相,检测波长为280nm,流速为每分钟0.6ml。分别取每1ml含蛋白质为12mg的人免疫球蛋白、人血白蛋白溶液各20μl,分别注入色谱柱,记录色谱图。人免疫球蛋白对照品单体峰与裂解体峰的分离度应大于1.5,人血白蛋白对照品单体峰与二聚体峰的分离度应大于1.5,拖尾因子按人血白蛋白单体峰计算应为0.95~1.40。
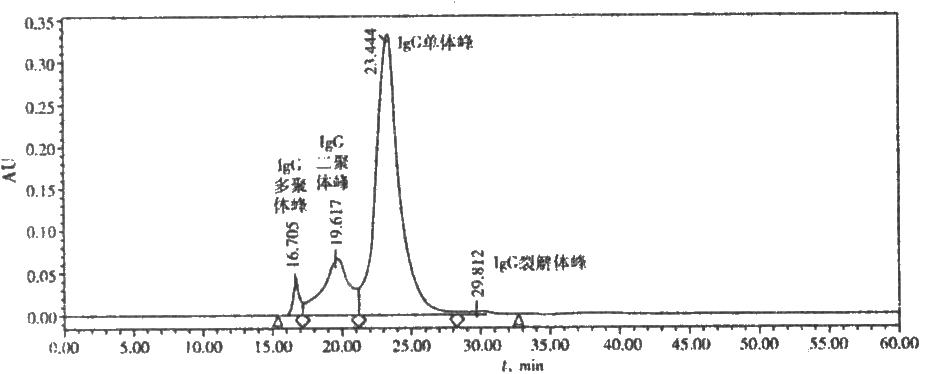
图 人免疫球蛋白IgG标准图谱
17.2 测定法
取供试品适量,用流动相稀释成每1ml约含蛋白质12mg的溶液,取20μl,注入色谱柱,记录色谱图60分钟。按面积归一法计算,色谱图中单体加二聚体峰的含量,即为IgG单体加二聚体含量。图谱各蜂的界限为两峰间最低点到基线的垂直线。主峰为IgG单体;相对保留时间约0.85的峰为二聚体。
18 附录Ⅵ S 人免疫球蛋白中甘氨酸含量测定法
本法系依据过量的6-氨基喹啉基-N-羟基琥珀酰亚氨基氨基甲酸酯(AQC)在一定条件下和氨基酸形成稳定的衍生产物(柱前衍生),用高效液相色谱法测定衍生产物,根据衍生产物的含量计算人免疫球蛋白中甘氨酸含量。
照高效液相色谱法(2010年版药典三部附录Ⅲ B)测定。
18.1 色谱条件与系统适应性试验
用十八烷基硅烷键合硅胶为基质的C18反相色谱柱,粒度4μm,内径3.9mm,柱长150mm;柱温为37℃;以140mmol/L醋酸钠、17mmol/L三乙胺(pH 5.65)、1μg/ml乙二胺四乙酸二钠为流动相A液,以100%乙腈为流动相B液,以纯水为流动相C液,流速为每分钟1.0ml,梯度洗脱32分钟(梯度表),检测波长为248nm。甘氨酸与相邻色谱峰之间分离度应大于1.5;拖尾因子(T)为0.95~1.40(甘氨酸和α-氨基丁酸峰);RSD应不大于2.0%(甘氨酸对照品峰面积测量值)。
18.2 内标溶液的制备
精密称取α-氨基丁酸对照品0.4g[1],加超纯水定容至100ml。
18.3 对照品溶液的制备
(1)精密称取甘氨酸对照品2.5g,加超纯水定容至100ml。
(2)精密量取(1)项溶液1.0ml,加9.0ml 1.5%磺基水杨酸,混匀静置2小时以上,以每分钟3000转离心10分钟,留取上清液备用。
(3)精密量取(2)项上清液0.4ml、0.8ml、1.0ml、1.2ml、1.6ml,分别置10ml量瓶中,用纯水定容。
(4)精密量取(3)项溶液各0.1ml,加纯水0.4ml,内标溶液0.02ml,混匀备用。
(5)精密量取(4)项溶液10μl放入衍生管中,加硼酸缓冲液(pH8~10)70μl涡旋混合,并加入20μl AQC衍生剂涡旋混合15秒,即为对照品溶液。
18.4 供试晶溶液的制备
(1)精密量取供试品溶液1.0ml,加1.5%磺基水杨酸9.0ml,混匀静置2小时以上,以每分钟3000转离心10分钟,留取上清液备用。
(2)精密量取(1)项上清液1.0ml,置10ml量瓶中,用纯水定容。
(3)精密量取(2)项溶液0.1ml,加0.4ml纯水,加内标溶液0.02ml,混匀后,精密量取10μl放入衍生管中加70μl硼酸缓冲液涡旋混合并加入20μl AQC衍生剂涡旋混合15秒,即为供试品溶液。
18.5 测定法
精密量取对照品溶液与供试品溶液,分别注入液相色谱仪,记录色谱图32分钟。进样量为10μl。按内标法计算。
梯度表
时间/分钟 | 流速/ml·min-1 | A液/% | B液/% | C液/% | 曲线 |
起始 | 1.0 | 100 | 0 | 0 | |
0.5 | 1.0 | 99.0 | 1.0 | 0 | 瞬时 |
18.00 | 1.0 | 95.0 | 5.0 | 0 | |
19.00 | 1.0 | 91.0 | 9.0 | 0 | |
22.00 | 1.0 | 83.0 | 17.0 | 0 | |
25.00 | 1.0 | 0 | 60.0 | 40.0 | 瞬时 |
28.00 | 1.0 | 100 | 0 | 0 | 瞬时 |
32.00 | 1.0 | 100 | 0 | 0 |
【附注】
(1)甘氨酸含量测定应采用柱前衍生及内标法,除本法要求外,衍生剂也可选用异硫氰酸苯酯、邻苯二甲醛;内标物也可选用正缬氨酸;C18反相色谱柱的粒度也可选用5μm或亚二微米。根据液相色谱系统、C18反相色谱柱规格、衍生剂及内标物的不同可以调整相应的色谱条件。
(2)直线回归相关系数应不低于0.999。
19 附录Ⅵ T 对羟基苯甲酸甲酯、对羟基苯甲酸丙酯含量测定法
本法系用气相色谱法测定供试品中对羟基苯甲酸甲酯及对羟基苯甲酸丙酯含量。
照气相色谱法(2010年版药典三部附录Ⅲ C)测定。
19.1 色谱条件与系统适用性试验
用涂布100%聚二甲基硅氧烷石英毛细管柱,柱温180℃,气化室温度250℃;氢离子化火焰检测器,检测器温度300℃。载气为氮气,流速为每分钟20ml。进样方式采用分流进样,进样量为1μl。内标物(对苯二酚)与对羟基苯甲酸甲酯及对羟基苯甲酸丙酯之间的分离度均应大于1.5,对羟基苯甲酸甲酯及对羟基苯甲酸丙酯的对照品溶液连续进样5次,所得对羟基苯甲酸甲酯及对羟基苯甲酸丙酯峰面积与对苯二酚峰面积之比的相对标准偏差应不大于5%。
19.2 内标溶液的制备
取对苯二酚50mg,精密称定,用无水乙醇定容至50ml,制成每1ml约含有1mg的内标溶液。
19.3 对照品溶液的制备
取对羟基苯甲酸甲酯0.1g、对羟基苯甲酸丙酯0.01g,精密称定,用无水乙醇定容至10ml,即得约1.00%对羟基苯甲酸甲酯的溶液、约0.10%对羟基苯甲酸丙酯的溶液。
19.4 校正因子测定用对照溶液的制备
取对照品溶液60μl,内标溶液100μl,加纯化水840μl,即得含内标物100μg/ml、对羟基苯甲酸甲酯约0.06%、对羟基苯甲酸丙酯约0.006%的校正因子测定用对照溶液。
19.5 测定法
取供试品840μl,加入内标溶液100μl、无水乙醇60μl,混匀,取1μl注入气相色谱仪,另取1μl校正因子测定用对照溶液,同法操作,按内标加校正因子测定法计算对羟基苯甲酸甲酯及对羟基苯甲酸丙酯含量。
20 附录Ⅵ U 重组人粒细胞刺激因子蛋白质含量测定法
本法采用高效液相色谱法测定供试品中重组人粒细胞刺激因子蛋白质含量。
照高效液相色谱法(2010年版药典三部附录Ⅲ B)测定。
20.1 色谱条件
色谱柱采用十八烷基硅烷键合硅胶为填充剂,孔径30nm,粒度5μm,直径4.6mm,长250mm;柱温为30℃±5℃,供试品保存温度为2~8℃;以0.1%三氟乙酸的水溶液为流动相A液,以0.1%三氟乙酸的乙腈溶液为流动相B液;流速为1ml/min;检测波长214nm;按下表进行梯度洗脱。
编号 | 时间/分钟 | A/% | B/% |
1 | 0 | 60 | 40 |
2 | 40 | 20 | 80 |
3 | 45 | 0 | 100 |
4 | 50 | 60 | 40 |
5 | 60 | 60 | 40 |
20.2 检查法
取1支标准品,按介绍复溶。用20mmol/L的醋酸-醋酸钠缓冲液(pH4.0)将标准品及供试品调节至相同蛋白质浓度,将供试品溶液与标准品溶液以相同
体积分别注入液相色谱仪(进样体积不小于10μl,进样量4~6μg),按上表进行梯度洗脱。标准品溶液、供试品溶液均进样3次,记录色谱图并计算峰面积。按下式计算重组人粒细胞刺激因子蛋白质含量(μg/ml):

nX为供试品溶液的稀释倍数。
21 附录Ⅵ V 残留溶剂测定法
药品中的残留溶剂系指在原料药或辅料的生产中,以及在制剂制备过程中使用的,但在工艺过程中未能完全去除的有机溶剂。药品中常见的残留溶剂及限度见附表1,除另有规定外,第一、第二、第三类溶剂的残留限度应符合附表1中的规定;对其他溶剂,应根据生产工艺的特点,制定相应的限度,使其符合产品规范、药品生产质量管理规范(GMP)或其他基本的质量要求。
本法照气相色谱法(2010年版药典三部附录Ⅲ C)测定。
21.1 色谱柱
21.1.1 1.毛细管柱
除另有规定外,极性相近的同类色谱柱之间可以互换使用。
(1)非极性色谱柱 固定液为100%的二甲基聚硅氧烷的毛细管柱。
(2)极性色谱柱 固定液为聚乙二醇(PEG-20M)的毛细管柱。
(3)中极性色谱柱 固定液为(35%)二苯基-(65%)甲基聚硅氧烷、(50%)二苯基-(50%)二甲基聚硅氧烷、(35%)二苯基-(65%)二甲基聚硅氧烷、(14%)氰丙基苯基-(86%)二甲基聚硅氧烷、(6%)氰丙基苯基-(94%)二甲基聚硅氧烷的毛细管柱等。
(4)弱极性色谱 柱固定液为(5%)苯基-(95%)甲基聚硅氧烷、(5%)二苯基-(95%)二甲基硅氧烷共聚物的毛细管柱等。
21.1.2 2.填充柱
以直径为0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他适宜的填料作为固定相。
21.2 系统适用性试验
(1)用待测物的色谱峰计算,毛细管色谱柱的理论板数一般不低于5000;填充柱的理论板数一般不低于1000。
(2)色谱图中,待测物色谱峰与其相邻色谱峰韵分离度应大于1.5。
(3)以内标法测定时,对照品溶液连续进样5次,所得待测物与内标物峰面积之比的相对标准偏差(RSD)应不大于5%;若以外标法测定,所得待测物峰面积的RSD应不大于10%。
21.3 供试品溶液的制备
21.3.1 1.顶空进样
除另有规定外,精密称取供试品0.1~1g,通常以水为溶剂;对于非水溶性药物,可采用N,N-二甲基甲酰胺、二甲基亚砜或其他适宜溶剂;根据供试品和待测溶剂的溶解度,选择适宜的溶剂且应不干扰待测溶剂的测定。根据各品种项下残留溶剂的限度规定配制供试品溶液,其浓度应满足系统定量测定的需要。
21.3.2 2.溶液直接进样
精密称取供试品适量,用水或合适的有机溶剂使溶解;根据各品种项下残留溶剂的限度规定配制供试品溶液,其浓度应满足系统定量测定的需要。
21.4 对照品溶液的制备
精密称取各品种项下规定检查的有机溶剂适量,采用与制备供试品溶液相同的方法和溶剂制备对照品溶液;如用水作溶剂,应先将待测有机溶剂溶解在50%二甲基亚砜或N,N-二甲基甲酰胺溶液中,再用水逐步稀释。若为限度检查,根据残留溶剂的限度规定确定对照品溶液的浓度;若为定量测定,为保证定量结果的准确性,应根据供试品中残留溶剂的实际残留量确定对照品溶液的浓度;通常对照品溶液色谱蜂面积不宜超过供试品溶液中对应的残留溶剂色谱蜂面积的2倍。必要时,应重新调整供试品溶液或对照品溶液的浓度。
21.5 测定法
21.5.1 第一法(毛细管柱顶空进样等温法)
当需要检查有机溶剂的数量不多,且极性差异较小时,可采用此法。
色谱条件 柱温一般为40~100℃;常以氮气为载气,流速为每分钟1.0~2.0ml;以水为溶剂时顶空瓶平衡温度为70~85℃,顶空瓶平衡时间为30~60分钟;进样口温度为200℃;如采用火焰离子化检测器(FID),温度为250℃。
测定法 取对照品溶液和供试品溶液,分别连续进样不少于2次,测定待测峰的峰面积。
对色谱图中未知有机溶剂的鉴别,可参考附表2进行初筛。
21.5.2 第二法(毛细管柱顶空进样系统程序升温法)
当需要检查的有机溶剂数量较多,且极性差异较大时,可采用此法。
色谱条件 柱温一般先在40℃维持8分钟,再以每分钟8℃的升温速率升至120℃,维持10分钟;以氮气为载气,流速为每分钟2.0ml;以水为溶剂时顶空瓶平衡温度为70~85℃,顶空瓶平衡时间为30~60分钟;进样口温度为200℃;,如采用FID检测器,进样口温度为250℃。具体到某个品种的残留溶剂检查时,可根据该品种项下残留溶剂的组成调整升温程序。
测定法 取对照品溶液和供试品溶液,分别连续进样不少于2次,测定待测峰的峰面积。
对色谱图中未知有机溶剂的鉴别,可参考附表3进行初筛。
21.5.3 第三法(溶液直接进样法)
可采用填充柱,亦可采用适宜极性的毛细管柱。
测定法 取对照品溶液和供试品溶液,分别连续进样2~3次,测定待测峰的峰面积。
21.6 计算法
(1)限度检查 除另有规定外;按各品种项下规定的供试品溶液浓度测定。以内标法测定时,供试品溶液所得被测溶剂峰面积与内标峰面积之比不得大于对照品溶液的相应比值。以外标法测定时,供试品溶液所得被测溶剂峰面积不得大于对照品溶液的相应峰面积。
(2)定量测定 按内标法或外标法计算各残留溶剂的量。
21.7 【附注】
(1)除另有规定外,顶空条件的选择:
①应根据供试品中残留溶剂的沸点选择顶空平衡温度。对沸点较高的残留溶剂,通常选择较高的平衡温度;但此时应兼顾供试品的热分解特性,尽量避免供试品产生的挥发性热分解产物对测定的干扰。
②顶空平衡时间一般为30~45分钟,以保证供试品溶液的气一液两相有足够的时间达到平衡。顶空平衡时间通常不宜过长,如超过60分钟,可能引起顶空瓶的气密性变差,导致定量准确性的降低。
(2)定量方法的验证 当采用顶空进样时,供试品与对照品处于不完全相同的基质中,故应考虑气液平衡过程中的基质效应(供试品溶液与对照品溶液组成差异对顶空气-液平衡的影响)。由于标准加入法可以消除供试品溶液基质与对照品溶液基质不同所致的基质效应的影响,故通常采用标准加入法验证定量方法的准确性;当标准加入法与其他定量方法的结果不一致时,应以标准加入法的结果为准。
(3)干扰峰的排除 供试品中的未知杂质或其挥发性热降解物易对残留溶剂的测定产生干扰。干扰作用包括在测定的色谱系统中未知杂质或其挥发性热降解物与待测物的保留值相同(共出峰);或热降解产物与待测物的结构相同(如甲氧基热裂解产生甲醇)。当测定的残留溶剂超出限度,但未能确定供试品中是否有未知杂质或其挥发性热降解物对测定有干扰作用时,应通过试验排除干扰作用的存在。对第一类干扰作用,通常采用在另一种极性不同的色谱柱系统中对相同供试品再进行测定,比较不同色谱系统中测定结果的方法。如两者结果一致,则可以排除测定中有共出峰的干扰;如两者结果不一致,则表明测定中有共出峰的干扰。对第二类干扰作用,通常要通过测定已知不含该溶剂的对照样品来加以判断。
(4)含氮碱性化合物的测定 普通气相色谱仪中的不锈钢管路、进样器的衬管等对有机胺等含氮碱性化合物具有较强的吸附作用,致使其检出灵敏度降低,应采用惰性的硅钢材料或镍钢材料管路;采用溶液直接进样法测定时,供试品溶液应不呈酸性,以免待测物与酸反应后不易汽化。
通常采用弱极性的色谱柱或其填料预先经碱处理过的色谱柱分析含氮碱性化合物,如果采用胺分析专用柱进行分析,效果更好。
对不宜采用气相色谱法测定的含氮碱性化合物,如N-甲基吡咯烷酮等,可采用其他方法如离子色谱法等测定。
(5)检测器的选择 对含卤素元素的残留溶剂如三氯甲烷等,采用电子捕获检测器(ECD),易得到高的灵敏度。
(6)由于不同的实验室在测定同一供试品时可能采用了不同的实验方法,当测定结果处于合格与不合格边缘时,以采用内标法或标准加入法为准。
(7)顶空平衡温度一般应低于溶解供试品所用溶剂的沸点10℃以下,能满足检测灵敏度即可;对于沸点过高的溶剂,如甲酰胺、2-甲氧基乙醇、2-乙氧基乙醇、乙二醇、N-甲基吡咯烷酮等,用顶空进样测定的灵敏度不如直接进样,一般不宜用顶空进样方式测定。
(8)利用保留值定性是气相色谱中最常用的定性方法。色谱系统中载气的流速、载气的温度和柱温等的变化都会使保留值改变,从而影响定性结果。校正相对保留时间(RART)只受柱温和固定相性质的影响,以此作为定性分析参数较可靠。应用中通常选用甲烷测定色谱系统的死体积(tO):
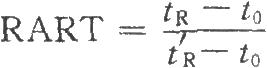
式中tR为组分的保留时间;
tR'为参比物的保留时间。
22 附表1 药品中常见的残留溶剂及限度
溶剂名称 | 限度/% |
第一类溶剂(应该避免使用) | |
苯 | 0.0002 |
四氯化碳 | 0.0004 |
0.0005 | |
0.0008 | |
0.15 | |
第二类溶剂(应该限制使用) | |
0.041 | |
0.036 | |
0.006 | |
0.388 | |
0.187 | |
0.06 | |
0.01 | |
N,N-二甲基乙酰胺 | 0.109 |
0.088 | |
二氧六环 | 0.038 |
溶剂名称 | 限度/% |
第二类溶剂(应该限制使用) | |
2-乙氧基乙醇 | 0.016 |
0.062 | |
甲酰胺 | 0.022 |
0.029 | |
0.3 | |
2-甲氧基乙醇 | 0.005 |
甲基丁基酮 | 0.005 |
0.118 | |
0.053 | |
0.005 | |
0.02 | |
0.016 | |
四氢化萘 | 0.01 |
四氢呋哺 | 0.072 |
甲苯 | 0.089 |
1,1,2-三氯乙烯 | 0.008 |
二甲苯① | 0.217 |
溶剂名称 | 限度/% |
第三类溶剂(药品GMP或其他质量要求限制使用) | |
0.5 | |
0.5 | |
甲氧基苯 | 0.5 |
0.5 | |
仲丁醇 | 0.5 |
乙酸丁酯 | 0.5 |
叔丁基甲基醚 | 0.5 |
异丙基苯 | 0.5 |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 |
溶剂名称 | 限度/% |
第三类溶剂(药品GMP或其他质量要求限制使用) | |
丁酮 | 0.5 |
甲基异丁基酮 | 0.5 |
异丁醇 | 0.5 |
0.5 | |
正戊醇 | 0.5 |
正丙醇 | 0.5 |
0.5 | |
乙酸丙酯 | 0.5 |
第四类溶剂(尚无足 够毒理学资料)② | |
1,1-二乙氧基丙烷 | |
1,1-二甲氧基甲烷 | |
异辛烷 | |
甲基异丙基酮 | |
甲基四氢呋喃 | |
三氯醋酸 | |
三氟醋酸 |
①通常含有60%间二甲苯、14%对二甲苯、9%邻二甲苯和17%乙苯。
②药品生产企业在使用时应提供该类溶剂在制剂中残留水平的合理性论证报告。
23 附表2 常见有机溶剂在等温法测定时相对于丁酮的保留值参考值
非极性色谱柱 | ||
溶剂名称 | tR/min | RART |
柱温40℃ | ||
正丙醇 叔丁基甲基醚 丁酮 仲丁醇 异丁醇 甲基异丙基酮 苯 四氯化碳 甲基四氢呋喃 二氧六环 异辛烷 乙酸丙酯 甲基异丁基酮 甲苯 正戊醇 | 1.828 2.090 2.179 2.276 2.356 2.487 2.489 2.522 2.584 2.609 2.635 2.655 2.807 2.982 3.109 3.252 3.449 3.666 3.898 3.908 3.913 3.954 4.264 4.264 4.517 4.808 4.976 4.985 5.281 5.311 5.340 5.470 5.583 5.676 6.760 6.823 6.957 7.434 7.478 8.628 8.738 8.870 9.283 11.180 11.382 1.54 | 0.126 0.268 0.315 0.368 0.411 0.481 0.482 0.501 0.534 0.547 0.561 0.572 0.654 0.748 0.817 0.894 1.000 1.117 1.242 1.247 1.250 1.272 1.439 1.440 1.576 1.733 1.823 1.828 1.988 2.004 2.019 2.089 2.150 2.201 2.785 2.819 2.891 3.148 3.172 3.792 3.851 3.922 4.145 5.168 5.276 |
柱温80℃ | ||
甲基丁基酮 乙酸丁酯 甲氧基苯 | 3.611 3.859 4.299 5.253 7.436 | 2.099 2.345 2.778 3.726 5.890 |
续表
极性色谱柱 | ||
溶剂名称 | tR/min | RART |
柱温40℃ | ||
异辛烷 叔丁基甲基醚 甲基环已烷 甲基四氢呋喃 四氯化碳 丁酮 甲基异丙基酮 苯 乙酸丙酯 甲基异丁基酮 仲丁醇 甲苯 正丙酵 二氧六环 乙酸丁酯 甲基丁基酮 | 1.682 1.787 1.842 1.926 1.943 2.005 2.021 2.159 2.209 2.243 2.405 2.876 2.967 3.000 3.347 3.403 3.481 3.635 3.6a3 3.810 3.980 4.062 4.079 4.604 4.716 4.758 4.822 4.975 4.977 6.020 6.643 7.202 7.368 7.497 7.985 8.390 8.746 9.238 10.335 10.827 11.012 11.486 1.602 | 0.032 0.075 0.097 0.131 0.138 0.163 0.169 0.225 0245 0.259 0.324 0.515 0.551 0.564 0.705 0.727 0.758 0.821 0.828 0.891 0.960 0.993 1.000 1.212 1.257 1.274 1.300 1.362 1.362 1.784 2.035 2.261 2.328 2.380 2.577 2.740 2.884 3.083 3.526 3.724 3.799 3.990 |
柱温80℃ | ||
异丁醇 异丙基苯 正戊醇 | 3.577 4.460 4.885 5.288 5.625 5.934 6.439 7.332 | 3.045 4.334 4.948 5.543 6.035 6.486 7.223 8.527 |
续表
24 附表3 常见有机溶剂在程序升温法测定时相对于丁酮的保留值参考值
非极性色谱柱 | ||
溶剂名称 | tR/min | RART |
正丙醇 叔丁基甲基醚 丁酮 仲丁醇 异丁醇 甲基异丙基酮 苯 四氯化碳 甲基四氢呋喃 异辛烷 | 1.846 2.121 2.201 2.303 2.401 2.512 2.519 2.544 2.611 2.623 2.665 2.674 2.839 3.051 3.128 3.302 3.507 3.756 3.966 3.971. 3.981 4.005 4.387 4.397 4.6124 4.843 5.087 5.099 5.380 5.398 5.402 5.501 5.649 5.739 6.815 6.928 | 0.127 0.272 0.314 0.367 0.419 0.477 0.481 0.494 0.529 0.535 0.558 0.562 0.649 0.760 0.801 0.892 1.000 1.131 1.241 1.244 1.249 1.262 1.462 1.468 1.581 1.702 1.830 1.837 1.984 1.994 1.996 2.048 2.126 2.173 . 2.738 2.798 |
续表
非极性色谱柱 | ||
溶剂名称 | tR/min | RART |
二氧六环 乙酸丙酯 甲基异丁基酮 甲苯 正戊醇 甲基丁基酮 乙酸丁酯 甲氧基苯 异丙基苯 四氢化萘 | 6.928 7.563 7.583 8.581 8.830 8.968 9.178 10.259 10.448 10.638 11.025 12.175 13.166 15.270 15.724 22.409 1.604 | 2.798 3.131 3.142 3.666 3.797 3.870 3.980 4.548 4.647 4.747 4.951 5.555 6.076 7.181 7.420 10.933 |
极性色谱柱 | ||
溶剂名称 | tR/min | RART |
异辛烷 叔丁基甲基醚 甲基四氢呋喃 四氯化碳 丁酮 甲基异丙基酮 苯 乙酸丙酯 甲基异丁基酮 仲丁醇 | 1.691 1.807 1.856 1.957 1.966 2.053 2.063 2.217 2.267 2.303 2.488 2.988 3.094 3.126 3.511 3.561 3.653 3.821 3.833 4.017 4.207 4.295 4.303 4.875 5.005 5.041 5.069 5.275 5.275 6.437 7.108 7.735 7.892 8.068 8.533 8.848 | 0.033 0.076 0.094 0.131 0.135 0.167 0.171 0.228 0.246 0.260 0.328 0.513 0.552 0.564 0.707 0.725 0.759 0.822 0.826 0.894 0.964 0.997 1.000 1.212 1.260 1.273 1.284 1.360 1.360 1.790 2.039 2.271 2.329 2.394 2.566 2.683 |
续表
极性色谱柱 | ||
溶剂名称 | tR/min | RART |
甲苯 正丙醇 二氧六环 乙酸丁酯 甲基丁基酮 异丁醇 异丙基苯 正戊醇 甲氧基苯 四氧化萘 | 9.156. 9.461 10.183 10.446 10.543 10.801 11.606 13.046 13.258 13.396 13.949 14.519 14.562 15.516 17.447 21.708 1.602 | 2.797 2.910 3.177 3.274 3.310 3.406 3.704 4.237 4.315 4.367 4.571 4.782 4.798 5.151 5.866 7.444 |
注:附表2、3中数据为非极性的SPB-1柱(30m×0.32mm,1.0μm)和极性的HP-INNOWAX柱(30m×0.32mm,0.5μm)测定的结果。
25 附录Ⅵ W 乙酰色氨酸测定法
本法系用紫外-可见分光光度法(吸收系数法)测定人血白蛋白供试品中的N-乙酰-DL-色氨酸含量。
25.1 测定法
用生理氯化钠溶液将供试品蛋白质稀释至5%,即为供试品溶液.量取供试品溶液0.1ml,分别加入生理氯化钠溶液0.3ml和0.3mol/L高氯酸溶液3.6ml,混匀;另取生理氯化钠溶液0.4ml,加0.3mol/L高氯酸溶液3.6ml,混匀,作为空白对照。室温放置10分钟,以每分钟3500转离。心20分钟,取上清液在波长280nm处测定吸光度,用空白溶液调零点。按下式计算供试品中的N-乙酰-DL-色氨酸含量:

式中n为供试品的稀释系数;
P为供试品的蛋白质含量,g/L。
26 附录Ⅵ X 氰化物残留量测定法
本法系依据在酸性条件下,溴化氰与吡啶联苯胺发生显色反应,采用紫外-可见分光光度法测定Hib多糖衍生物中溴化氰的含量。
26.1 试剂
(3)吡啶联苯胺溶液 精密称定联苯胺0.5g,加入60%吡啶溶液50ml溶解,再加入2%盐酸10ml,混匀。临用前配制。
26.2 对照品溶液的制备
(1) 0.1mg/ml溴化氰对照品贮备液 称定溴化氰10mg,加水定容至100ml(乙腈助溶)。临用前配制。
(2)溴化氰对照品工作液(500ng/ml) 精密量取溴化氰对照品贮备液1ml,加水定容至200ml,混匀。
26.3 供试品溶液的制备
26.4 测定法
量取吡啶联苯胺溶液2.0ml,加水2.0ml,混匀,20℃以下、暗处放置15分钟后,在波长520nm处测定吸光度,作为空白对照。
量取供试品溶液2.0ml、吡啶联苯胺溶液2.0ml,混匀,20℃以下、暗处放置15分钟后,在波长520nm处测定吸光度。
分别量取溴化氰对照品工作液0.1ml、0.2ml、0.4ml、0.6ml、0.8ml、1.0ml于试管中,每管依次加水1.9ml、1.8ml、1.6ml、1.4ml、1.2ml、1.0ml,加入吡啶联苯胺溶液2.0ml,混匀,20℃以下、暗处放置15分钟后,在波长520nm处测定吸光度。
26.5 结果计算
以对照品工作液的含量对其相应的吸光度作直线回归,求得直线回归方程,将供试品溶液的吸光度代入直线回归方程,求出供试品溶液中溴化氰的含量B(ng/ml)。
供试品中溴化氰的含量(ng/mg)=B/20
式中 B为供试品溶液中溴化氰的含量,ng/ml;
27 附录Ⅵ Y 碳二亚胺残留量测定法
本法系依据二甲基巴比妥酸试液与碳二亚胺(EDAC)反应形成紫红色络合物,采用紫外-可见分光光度法测定碳二亚胺的含量。
27.1 试剂
(1)二甲基巴比妥酸试液 称取二甲基巴比妥酸1g于16ml吡啶中,并加水至20ml.混匀,临用现配。
(2)醋酸吡啶溶液 将等体积的冰醋酸和吡啶混匀制成,临用现配。
(3)100μmol/L EDAC对照品贮备液 称取0.0192gEDAC,以水溶解并定容至100ml,得1mmol/L溶液,量取1mmol/L溶液1ml于10ml量瓶,加水定容至刻度,即得100μmol/L EDAC对照品贮备液。临用现配。(4)EDAC对照品工作液的制备 取100μmol/LEDAC对照品贮备液,用水分别稀释至10μmol/L、20μmol/L、 40μmol/L、 60μmol/L、 80μmol/L, 即为EDAC对照品工作液。
27.2 测定法
取供试品和EDAC对照品工作液各0.2ml,分别加入二甲基巴比妥酸试液1.8ml,另取水0.2ml作为空白对照,同法操作,混匀各管,室温暗处静置30分钟,分别加入醋酸吡啶溶液2.0ml,混匀后在波长599nm处测定吸光度(如试验有干扰,在测吸光度前以每分钟4000转离心5分钟)。
27.3 结果计算
以EDAC对照品溶液的浓度对其相应的吸光度作直线回归,求得直线回归方程。将供试品溶液的吸光度代入直线回归方程,求出含量,取其平均值。供试品EDAC残留量(μmol/L)=供试品溶液EDAC的平均浓度×稀释倍数
28 参考资料
- ^ [1] 国家药典委员会.中华人民共和国药典:2010年版:第一增补本[M].北京:中国医药科技出版社,2010.